Mass Spectrometry
Explore Poochon’s Proteomics Solutions
With accumulated experience and expertise in the biological application of mass spectrometry, Poochon’s scientists developed a variety of mass spectrometry-based protocols and workflows for analysis of proteins and proteomes from any biological sample to support different applications. Three fundamental platforms were developed by our scientists.
PROMICFINDER Platform for Protein Identification and Quantification
PROMICFINDER, from Poochon, is a proteomic technology capable of identifying and measuring rapidly (high throughput), broadly (thousands of proteins simultaneously), and deeply (high-and low-abundance proteins).
It is a highly sensitive, quantitative, and reproducible proteomic tool for profiling up to 3,000 different proteins from a single protein gel band sample, one IP sample, or a single un-fractioned cell/tissue lysate sample by a 110-minute LC-MS/MS run.
- Post-translational modifications can be simultaneously identified. PTMs such as phosphorylation (S/T/Y), acetylation (K), ubiquitination (K), and O-linked/N-linked glycans can be identified from both IP and lysate samples. For specific target PTMs, the enrichment of target proteins, for example by IP, is generally required.
- Our SOPs are designed to accommodate protein extraction from any biological sample to ensure data quality and reproducibility.
- Examples: IP, pull-down, cellular fraction, cellular organelle, cell pellet or lysate, tissue pellet or tissue lysate, and bodily fluid
- Results are reported in a standardized format ready for generating publishable data. The standard deliverables include: the study report in PDF file format, MS data in Excel file format, and related figures in PowerPoint file format.
- SPECIFICITY – FDR (false discovery rate) is smaller than 1%, MS1 delta mass tolerance is 15 ppm and MS2 delta mass is less than 0.02 Da.
- REPRODUCIBILITY – PROMICFINDER, using LC-MS/MS for protein identification and quantification obtained data, achieves coefficients of variation much lower than industry standards and can be seamlessly used for any research project consistently over time. CV is typically less than 10%.
- DYNAMIC RANGE – Quantitatively identifies proteins of very high abundance and very low abundance with a dynamic range up to 5 logs from 20 μg of protein lysate prepared from cell or tissue.
- SENSITIVITY – LLOD (lower limit of detection) of purified monoclonal antibody is 0.1 ng. Up to 3,000 different proteins can be consistently identified by analyzing tryptic peptides from 10 μg of protein lysate prepared from cell or tissue without sample pre-treatment by one 110-minute LC-MS/MS run. Protein Detection limit is at the 1 nano-gram level; PTM detection limit is generally at the 100 nano-gram level.
PROMICMAPPER Platform for Quantitative Proteome Profiling
Poochon developed a standardized platform, PROMICMAPPER, to measure changes in global protein expression in biological systems upon perturbations using isobaric mass tags (tandem mass tags (TMT) multiplex labeling kit) and Liquid Chromatography tandem-Mass Spectrometry (LC-MS/MS). PROMICMAPPER allows for quantitative protein profiling of up to 9,000 proteins for one project, or deep quantitative protein profiling of up to 12,000 proteins for one project.
Additionally, the data generated by PROMICMAPPER can be analyzed using Poochon’s PROMICPATH data and pathway analysis tool, which uses ten groups of housekeeping proteins/complexes to evaluate the completeness and variability of a proteome profile. The evaluation ensures the integrity, reliability, and reproducibility of a large set of protein profile data generated by PROMICMAPPER.
PROMICMAPPER is the most cost-efficient approach for identification of differential expression of proteins for elucidating the molecular mechanisms of perturbations and discovery of biomarkers.
- Quantitative protein profiling (up to 9,000 proteins) and deep quantitative protein profiling (up to 12,000 proteins)
- Cost-efficient approach for global protein profiling, proteome characterization, post-translational modifications profiling, biomarker discovery, identification of therapeutic or diagnostic markers in different diseases, identification of key regulators in major biological pathways, and validation of previous discoveries
- Standardized SOPs for sample preparation and LC-MS/MS analysis to ensure quality data and reproducibility
- Our SOPs are designed to accommodate protein extraction from any biological sample to ensure data quality and reproducibility.
- Examples: cell pellet or lysate, tissue pellet or tissue lysate, or bodily fluid
- Results are reported in a standardized format ready for generating publishable data. The standard deliverables include: the study report in PDF file format, MS data in Excel file format with statistical analysis and volcano plots, heat maps in R, and related figures in PowerPoint file format.
- PROTEOME COVERAGE – Up to 9,000 unique different proteins can be consistently quantified from one set of 18 human origin samples by using one set of TMT-pro 18plex.
- DYNAMIC RANGE – Quantitatively identifies proteins of very high abundance and very low abundance with a dynamic range up to 5 logs from 50 μg of protein lysate prepared from cell or tissue.
- SPECIFICITY – FDR (false discovery rate) is smaller than 1%, MS1 delta mass tolerance 15 ppm and MS2 delta mass is less than 0.02 Da.
- REPRODUCIBLE – PROMICMAPPER, using LC-MS/MS for protein identification and quantification obtained data, achieves coefficients of variation much lower than industry standards and can be seamlessly used for any research project consistently over time. R2 value (correlation) for the abundance of all identified proteins between sample replicates is typically larger than 0.99.
PROMICPATH for Proteomic Data Validation and Biological Pathway Analysis
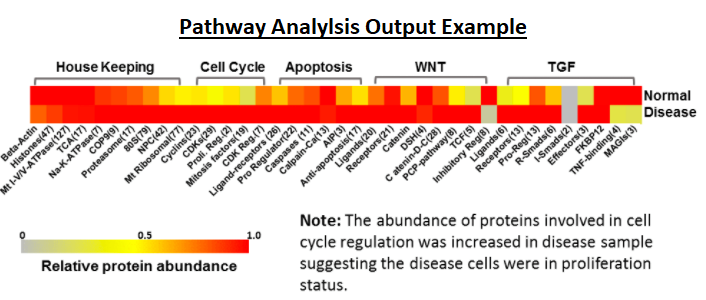
Poochon developed an “in-house” bioinformatic tool, PROMICPATH software, for analysis of large mass spectrometry proteomics datasets. PROMICPATH is designed for evaluation of the completeness and variability of a proteome profile, ensuring the integrity, reliability, and reproducibility of a large set of data generated, and biological pathway analysis of large mass spectrometry datasets.
PROMICPATH uses a pathway protein database established based on the KEGG pathway database (http://www.genome.jp/kegg/pathway.html), UniProtKB protein database and NCBI protein database. The “proteomics” datasets and differential expression protein profile is analyzed against this database.
For each pathway, the output includes four results:
1) Coverage: percentage of proteins identified in the pathway
2) Change: percentage of identified proteins significantly altered in abundance in the pathway
3) Up-Change: percentage of identified proteins significantly increased in abundance in the pathway
4) Down-Change: percentage of identified proteins significantly decreased in abundance in the pathway
The higher the percentage of changed proteins in a pathway indicates a higher possibility of pathway malfunction. Pathway analysis is available for human and mouse origin samples.
- Protein abundance variation and protein function grouping based pathway analysis algorithms
- Housekeeping proteins and complex based evaluation procedure ensures the integrity, reliability, and reproducibility of a large set of proteomic data (e.g. generated by PROMICMAPPER platform)
- Both common pathway analysis services and customized pathway analysis services
1. Housekeeping Proteins and Complexes
2. Cell cycle checkpoint
3. Apoptosis evaluation
4. Chromatin Modification
5. DNA replication and Repair
6. Transcriptional Regulation
7. Translation
8. Folding-sorting-degradation
9. Extracellular Matrix – Matrisome
10. Cellular Community
11. Cell motility-Regulation of actin cytoskeleton
12. PI3K/AKT
13. RAS
14. Notch
15. Hedgehog
16. MAPK
17. STAT/JAK
18. TGF
19. WNT
20. HIPPO
21. NF-kappa pathway
22. GPCR signaling pathway
23. p53 Pathway
24. Calcium Signaling
25. Nucleotide Metabolism
26. Amino Acid metabolism
27. Carbohydrate metabolism
28. Lipid metabolism
29. Energy Metabolism
30. Transport and catabolism
31. Warburg Effect
32. Excretory system
33. Digestive system
34. Heat Shock Protein Pathway
35. Necroptosis-pathway
36. Angiogenesis
37. Epithelial Mesenchymal Transition (EMT)
38. Signaling pathways regulating pluripotency of stem cells
39. Autophagy Regulation
40. Ubiquitination
41. ER-related pathway
42. Free Radicals